

{kind=link}
![]()
Determine 1: CoarsenConf structure.
Molecular conformer era is a basic process in computational chemistry. The target is to foretell steady low-energy 3D molecular buildings, generally known as conformers, given the 2D molecule. Correct molecular conformations are essential for numerous functions that rely upon exact spatial and geometric qualities, together with drug discovery and protein docking.
We introduce CoarsenConf, an SE(3)-equivariant hierarchical variational autoencoder (VAE) that swimming pools data from fine-grain atomic coordinates to a coarse-grain subgraph degree illustration for environment friendly autoregressive conformer era.
Background
Coarse-graining reduces the dimensionality of the issue permitting conditional autoregressive era relatively than producing all coordinates independently, as carried out in prior work. By immediately conditioning on the 3D coordinates of prior generated subgraphs, our mannequin higher generalizes throughout chemically and spatially comparable subgraphs. This mimics the underlying molecular synthesis course of, the place small practical models bond collectively to type giant drug-like molecules. Not like prior strategies, CoarsenConf generates low-energy conformers with the flexibility to mannequin atomic coordinates, distances, and torsion angles immediately.
The CoarsenConf structure will be damaged into the next parts:
(I) The encoder $q_phi(z| X, mathcal{R})$ takes the fine-grained (FG) floor fact conformer $X$, RDKit approximate conformer $mathcal{R}$ , and coarse-grained (CG) conformer $mathcal{C}$ as inputs (derived from $X$ and a predefined CG technique), and outputs a variable-length equivariant CG illustration by way of equivariant message passing and level convolutions.
(II) Equivariant MLPs are utilized to study the imply and log variance of each the posterior and prior distributions.
(III) The posterior (coaching) or prior (inference) is sampled and fed into the Channel Choice module, the place an consideration layer is used to study the optimum pathway from CG to FG construction.
(IV) Given the FG latent vector and the RDKit approximation, the decoder $p_theta(X |mathcal{R}, z)$ learns to get better the low-energy FG construction via autoregressive equivariant message passing. The whole mannequin will be educated end-to-end by optimizing the KL divergence of latent distributions and reconstruction error of generated conformers.
MCG Process Formalism
We formalize the duty of Molecular Conformer Era (MCG) as modeling the conditional distribution $p(X|mathcal{R})$, the place $mathcal{R}$ is the RDKit generated approximate conformer and $X$ is the optimum low-energy conformer(s). RDKit, a generally used Cheminformatics library, makes use of an inexpensive distance geometry-based algorithm, adopted by a reasonable physics-based optimization, to attain affordable conformer approximations.
Coarse-graining
![]()
Determine 2: Coarse-graining Process.
(I) Instance of variable-length coarse-graining. Superb-grain molecules are break up alongside rotatable bonds that outline torsion angles. They’re then coarse-grained to scale back the dimensionality and study a subgraph-level latent distribution. (II) Visualization of a 3D conformer. Particular atom pairs are highlighted for decoder message-passing operations.
Molecular coarse-graining simplifies a molecule illustration by grouping the fine-grained (FG) atoms within the unique construction into particular person coarse-grained (CG) beads $mathcal{B}$ with a rule-based mapping, as proven in Determine 2(I). Coarse-graining has been broadly utilized in protein and molecular design, and analogously fragment-level or subgraph-level era has confirmed to be extremely beneficial in various 2D molecule design duties. Breaking down generative issues into smaller items is an strategy that may be utilized to a number of 3D molecule duties and supplies a pure dimensionality discount to allow working with giant advanced programs.
We notice that in comparison with prior works that target fixed-length CG methods the place every molecule is represented with a set decision of $N$ CG beads, our technique makes use of variable-length CG for its flexibility and skill to help any selection of coarse-graining approach. Which means a single CoarsenConf mannequin can generalize to any coarse-grained decision as enter molecules can map to any variety of CG beads. In our case, the atoms consisting of every linked part ensuing from severing all rotatable bonds are coarsened right into a single bead. This selection in CG process implicitly forces the mannequin to study over torsion angles, in addition to atomic coordinates and inter-atomic distances. In our experiments, we use GEOM-QM9 and GEOM-DRUGS, which on common, possess 11 atoms and three CG beads, and 44 atoms and 9 CG beads, respectively.
SE(3)-Equivariance
A key side when working with 3D buildings is sustaining acceptable equivariance.
Three-dimensional molecules are equivariant beneath rotations and translations, or SE(3)-equivariance. We implement SE(3)-equivariance within the encoder, decoder, and the latent area of our probabilistic mannequin CoarsenConf. Consequently, $p(X | mathcal{R})$ stays unchanged for any rototranslation of the approximate conformer $mathcal{R}$. Moreover, if $mathcal{R}$ is rotated clockwise by 90°, we anticipate the optimum $X$ to exhibit the identical rotation. For an in-depth definition and dialogue on the strategies of sustaining equivariance, please see the total paper.
Aggregated Consideration
![]()
Determine 3: Variable-length coarse-to-fine backmapping by way of Aggregated Consideration.
We introduce a technique, which we name Aggregated Consideration, to study the optimum variable size mapping from the latent CG illustration to FG coordinates. It is a variable-length operation as a single molecule with $n$ atoms can map to any variety of $N$ CG beads (every bead is represented by a single latent vector). The latent vector of a single CG bead $Z_{B}$ $in R^{F instances 3}$ is used as the important thing and worth of a single head consideration operation with an embedding dimension of three to match the x, y, z coordinates. The question vector is the subset of the RDKit conformer similar to bead $B$ $in R^{ n_{B} instances 3}$, the place $n_B$ is variable-length as we all know a priori what number of FG atoms correspond to a sure CG bead. Leveraging consideration, we effectively study the optimum mixing of latent options for FG reconstruction. We name this Aggregated Consideration as a result of it aggregates 3D segments of FG data to type our latent question. Aggregated Consideration is answerable for the environment friendly translation from the latent CG illustration to viable FG coordinates (Determine 1(III)).
Mannequin
CoarsenConf is a hierarchical VAE with an SE(3)-equivariant encoder and decoder. The encoder operates over SE(3)-invariant atom options $h in R^{ n instances D}$, and SE(3)-equivariant atomistic coordinates $x in R^{n instances 3}$. A single encoder layer consists of three modules: fine-grained, pooling, and coarse-grained. Full equations for every module will be discovered within the full paper. The encoder produces a last equivariant CG tensor $Z in R^{N instances F instances 3}$, the place $N$ is the variety of beads, and F is the user-defined latent measurement.
The position of the decoder is two-fold. The primary is to transform the latent coarsened illustration again into FG area via a course of we name channel choice, which leverages Aggregated Consideration. The second is to refine the fine-grained illustration autoregressively to generate the ultimate low-energy coordinates (Determine 1 (IV)).
We emphasize that by coarse-graining by torsion angle connectivity, our mannequin learns the optimum torsion angles in an unsupervised method because the conditional enter to the decoder will not be aligned. CoarsenConf ensures every subsequent generated subgraph is rotated correctly to attain a low coordinate and distance error.
Experimental Outcomes
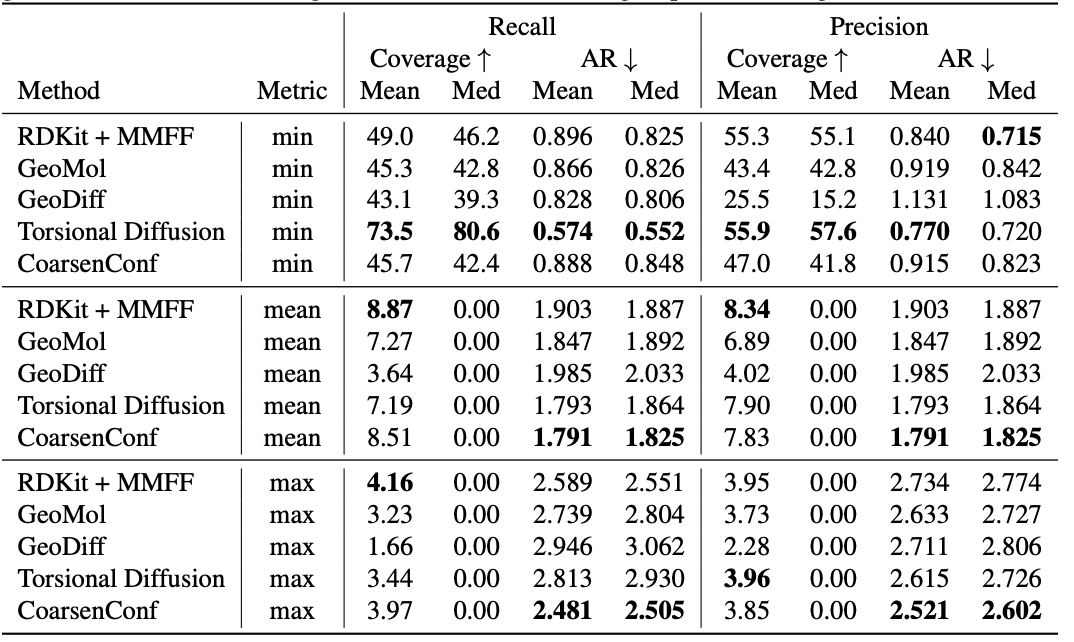
Desk 1: High quality of generated conformer ensembles for the GEOM-DRUGS take a look at set ($delta=0.75Å$) by way of Protection (%) and Common RMSD ($Å$). CoarsenConf (5 epochs) was restricted to utilizing 7.3% of the information utilized by Torsional Diffusion (250 epochs) to exemplify a low-compute and data-constrained regime.
The common error (AR) is the important thing metric that measures the common RMSD for the generated molecules of the suitable take a look at set. Protection measures the proportion of molecules that may be generated inside a particular error threshold ($delta$). We introduce the imply and max metrics to raised assess sturdy era and keep away from the sampling bias of the min metric. We emphasize that the min metric produces intangible outcomes, as except the optimum conformer is understood a priori, there isn’t any solution to know which of the 2L generated conformers for a single molecule is greatest. Desk 1 reveals that CoarsenConf generates the bottom common and worst-case error throughout the whole take a look at set of DRUGS molecules. We additional present that RDKit, with a reasonable physics-based optimization (MMFF), achieves higher protection than most deep learning-based strategies. For formal definitions of the metrics and additional discussions, please see the total paper linked under.
For extra particulars about CoarsenConf, learn the paper on arXiv.
BibTex
If CoarsenConf evokes your work, please think about citing it with:
@article{reidenbach2023coarsenconf,
title={CoarsenConf: Equivariant Coarsening with Aggregated Consideration for Molecular Conformer Era},
writer={Danny Reidenbach and Aditi S. Krishnapriyan},
journal={arXiv preprint arXiv:2306.14852},
12 months={2023},
}